


Ostepetrosis Guide
This guide to the disease was written by Dr. Colin Steward, now retired Consultant in Paediatric Stem Cell Transplantation at the Bristol Royal Hospital for Children. The opinions expressed are personal and do not necessarily represent those of the Osteopetrosis Support Trust. The Trust can not offer medical advice to families. What is provided here should be used as a reference only, as every child is affected differently.
Although great care has been taken in the compilation and preparation of all entries to ensure accuracy, we cannot accept responsibility for any errors or omissions. Any medical information provided is for education/information purposes and is not designed to replace medical advice by a qualified medical professional.
What Is Osteopetrosis?
Osteopetrosis is a term used to describe a range of rare genetic diseases in which the density of bones is increased. This is the opposite of what is seen in the common bone thinning condition, Osteoporosis, where bone density is too low.
Osteopetrosis results from problems in formation or function of Osteoclasts, cells which live in bone and dissolve solid bone so that it can be rebuilt and reshaped during growth or repaired after injury.
In the most severe forms of Osteopetrosis children are diagnosed soon after birth, but in others diagnosis may only be made when an X-ray is taken to investigate a broken bone as an adult. Many of the forms which cause disease in babies are curable by blood or marrow stem cell transplantation but no cure has yet been developed for the later onset (properly called Autosomal Dominant Osteopetrosis, ADO)
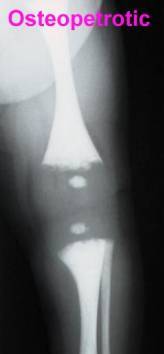
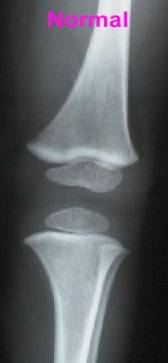
The picture on the left shows an X-ray of the leg bones in a baby with osteopetrosis. In comparison to the more normal appearances on the right (in a child who has undergone successful bone marrow transplantation) the bones are a dense, chalky white colour. This explains the alternative name of “Marble Bone Disease”.
Osteoclasts & What They Do
At every stage of life we need to mould and strengthen our bones so that they:
1. Stay strong but light so as to be best at resisting stresses and healing fractures
Most bones have a hollow or tubular structure in order to maximise their ratio of strength to weight.
2. Grow as a child grows
It is extremely important that the rate of growth of bones keeps up with the growth of the rest of the body. Bones need to be hollowed out from the centre and grow at the edges if they are to get bigger but stay light. This "remodeling" of bone is especially important in the skull where nerves pass through small holes (called foramina) to the eyes, ears and muscles of the face, and where the spinal nerves pass through the bottom of the skull into the spine. If these holes do not grow at the same rate as the nerves then the bone exerts pressure on the nerves (or the veins and arteries supplying them with blood) and this can damage them. Problems which occur as a result include visual loss, deafness, facial nerve paralysis and hydrocephalus (excessive fluid around the brain).
3. House the bone marrow
The bone marrow, the factory for the blood, is contained in the cavities in the centre of bones. If these cavities cannot form properly, blood is produced mainly in the liver and spleen - the same as occurs in a young baby in the womb. This causes swelling of the liver and spleen, resulting in abdominal swelling, and can strangely in turn lead to excessive destruction of blood cells, low red cell and platelet counts, and need for transfusions.

TYPES OF OSTEOPETROSIS
TYPES OF OSTEOPETROSIS (click to see relevant section)
The disease occurs in a variety of forms, listed here in approximate order of severity and beginning with the mildest:
Transient Infantile Osteopetrosis
This is the least reported form of the disease and resolves without intervention. It may represent a mild form of the disease. The importance of knowing of this disease subtype is that it highlights the need to perform a check X-ray in any child about to undergo bone marrow transplantation just before chemotherapy commences. This will reduce the risk (although very small) of transplanting a child with self-resolving osteopetrosis.
Adult, "benign", Autosomal Dominant Osteopetrosis (ADO)
This was the first form of the disease to be described, by a German radiologist called Albers-Schönberg in 1904, and occurs in two forms:
* ADO Type I which is typified by thickening (sclerosis) of the dome of the skull (skull vault)
This rare form of the diseases is caused by mutations of the LRP5 gene.
* ADO Type 2 (ADO2, classical Albers-Schönberg disease) where the spine has a so-called "rugger jersey" striped appearance and the pelvic bones contain endobones.
This is the commonest form of the disease and is usually diagnosed in adolescence or early adulthood. Estimates suggest that up to 5.5 of every 100,000 people are affected. Inheritance is autosomal dominant meaning that there one or other parent of the person with osteopetrosis may have had the disease themselves, although the disease may vary in severity between generations of the family.
The most common symptoms are an increased risk of fracture, back / bone pain, headache and osteomyelitis (infection of the bone). Problems due to nerve compression (deafness, visual loss and facial nerve paralysis) occur much more rarely than in the severe infant forms, affecting only approximately one in six patients. 80% of patients develop clinical problems due to the disease. Fractures develop in almost all patients; these are often slow to heal. Osteomyelitis of the jaw or other bones affected approximately one quarter of those affected.
The gene responsible for most cases of ADO2 has been pinpointed as the ClCN7 chloride channel gene. This is the same gene that causes severe osteopetrosis in approximately 15% of children with osteopetrosis. The difference is that both maternal and paternal copies of the gene are affected in the children, but only one copy in adults with ADO2.
Stem cell transplantation has never been attempted in this form of the disease and there are currently no established, effective modes of therapy. Some recent work on a mouse model of the disease has led to the suggestion that interferon gamma-1b may play a role in reducing bone mass in this disease, as previously suggested in children with severe infantile osteopetrosis.
Carbonic Anhydrase type II (CAII) Deficiency
This disease is caused by a deficiency of an enzyme, CAII, which has activity in bones, kidneys and the brain, and all of these organs are therefore affected. It is rare and principally affects children of Mediterranean and Arab race. The gene responsible for producing CAII osteopetrosis is found on chromosome 8 (at position 8q22) and is inherited in autosomal recessive fashion. As well as causing increased bone density and a tendency to fracture easily, there are characteristic changes in body chemistry. The blood is slightly acidic and has a high chloride concentration ("hyperchloraemic acidosis"). The blood acidity is caused by excessive leakage of bicarbonate from the kidney tubules (termed renal tubular acidosis). CAII must also have an important role in the brain and affected children therefore develop progressive cerebral calcification and learning difficulties. Growth failure and dental malocclusion are other problems.
This disease usually causes symptoms in the first few years of life although X-rays are normal at birth. X-ray appearances often improve again in later life. Approximately three fifths of children develop nerve compression where nerves pass through the skull: this most commonly causes blindness, but can also result in hearing loss and facial palsy. Unlike malignant infantile osteopetrosis described below blood problems tend to be minor or absent.
Treatment with oral bicarbonate supplements does not seem to change the course of the disease. Bone marrow transplantation normalises bone density by providing healthy osteoclasts, but does not seem to prevent cerebral calcification and intellectual decline.
This disease should be excluded in every child with osteopetrosis, at the very least by scrutiny of blood bicarbonate and chloride and urine pH, but preferably by measuring CAII activity. This is particularly important in children of Mediterranean or Arab race and in children lacking blood derangement.
Pycnodysostosis
Pycnodysostosis is an autosomal recessive disease resulting from deficiency of an important enzyme in osteoclasts called cathepsin K. It causes short stature, short bones at the ends of the fingers (called distal phalanges) and delayed closure of the soft spot (fontanelle) on the top of the skull. The famous French artist Henri de Toulouse-Lautrec is thought to have had this disease.
Pycnodysostosis can be easy to confuse with other types of osteopetrosis in young babies when some of the most characteristic X-ray changes have not developed. It is not generally thought to be treatable by stem cell transplantation.
Malignant Infantile Osteopetrosis (MIOP)
This is the commonest of the severe forms of infantile osteopetrosis and is caused by faults in at least seven different genes. Most children develop severe nerve damage early in life and two thirds of affected children die by the age of 6 years without treatment. Currently the only curative treatment is blood or bone marrow stem cell transplantation. This can completely prevent further progression of the disease in most children, although in a small percentage of cases the bones do not respond.
It is now known that mutation in any one of seven genes is responsible for three quarters of cases, with no causative gene having so far been identified in the remaining quarter. The genes which may be changed include the proton pump (TCIRG1), chloride channel (ClCN7) OSTM1, RANK (TNFRSF11A), RANK Ligand (TNFSF11), SNX10, PLEKHM1 and NF-kB Essential Modulator (NEMO) genes.
TCIRG1 mutations account for approximately 50% of cases in young children, ClCN7 mutations 15% of cases, and each of SNX10, OSTM1, TNFRSF11A and TNFSF11 accounts for 2-4% of cases. Sequencing the gene responsible in each child has previously been a protracted process, especially in those who do not have mutations of either TCIRG1 or ClCN7. Fortunately it is now possible to sequence all of these genes simultaneously, as well as many other genes which cause osteosclerosis, by a technique called Next Generation sequencing or Massive Parallel Sequencing.
Neuronopathic Osteopetrosis
The most severe form of osteopetrosis causes severe progressive cerebral deterioration which seems to be due to abnormal underlying biochemistry in the brain rather than bone compression as in most other forms of osteopetrosis. Babies with this form of the disease often develop spasticity in the first few months of life and rarely live beyond the age of 1-2 years.
It is now known that many children with this form of osteopetrosis have mutation of a gene called OSTM1. This is a particularly common form of the disease in Bedouin Arab races, especially in the Kuwaiti population.
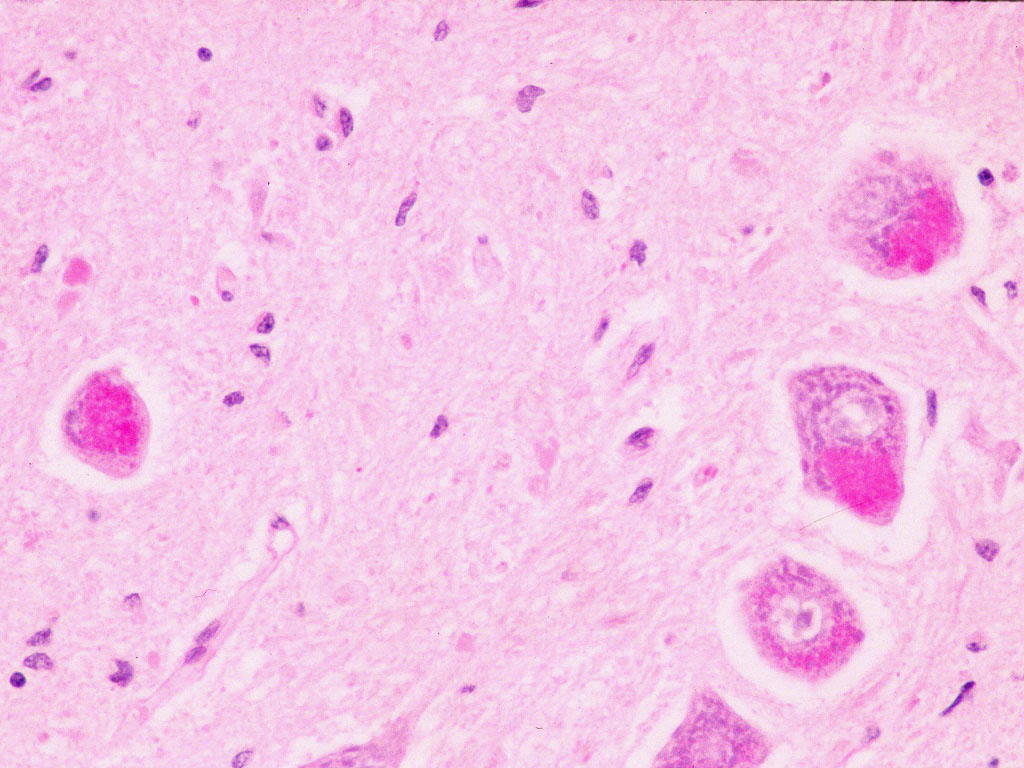
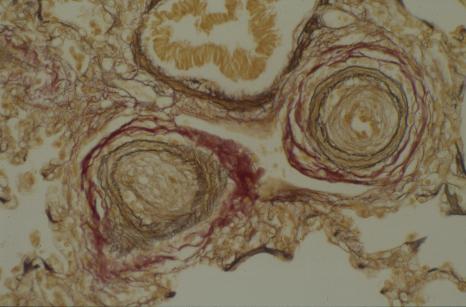
Biopsy of brain or spinal cord shows "inclusion bodies" (bright pink in the picture below) similar to those seen in a group of incurable neurodegenerative diseases, the ceroid lipofuscinoses, (also known as Batten disease). It is extremely important to recognise this form of the disease since it is completely incurable and blood/marrow stem cell transplantation is contraindicated.
SYMPTOMS OF OSTEOPETROSIS
This section refers mostly to the symptoms which develop in children with these severe (infantile malignant) form of the disease that is usually diagnosed in babies. Older children and adults with the autosomal dominant form of the disease may develop some of these symptoms to a milder degree but this is much more variable and unpredictable.
The commonest symptoms in order of frequency are:
Snuffles
Nasal congestion and discharge start very early in life and are generally constant. This is caused by overgrowth of the upper nasal bones and not usually, as some think, by persistent viral infections.
Impaired vision
This is the commonest symptom leading to medical diagnosis and is typically apparent by the second to fourth month of life. Approximately 75% of children with malignant infantile osteopetrosis will develop visual impairment, usually within the first year of life.
In many children optic nerve damage becomes apparent at around 6-8 weeks when they would usually begin to fix and follow faces, lights and large objects. Instead the eyes begin to scan rapidly from side to side resulting in wobbly movements called nystagmus. An example of nystagmus, although not from a baby with osteopetrosis, is shown in this video.
The optic nerve carries impulses from the retina at the back of the eye to the brain and passes through a bony optic canal in the skull on its way from the eye socket to the brain. This can be seen on computerised tomography (CT) scans. One way of looking at these scans is by 3D computer reconstructions. In the scan below we are looking at the eye sockets of a patient from the front. The opening of the optic canal, the optic foramen, can be seen at the rear of the left socket as a small black spot. It is narrowing at this point that can damage the nerve.
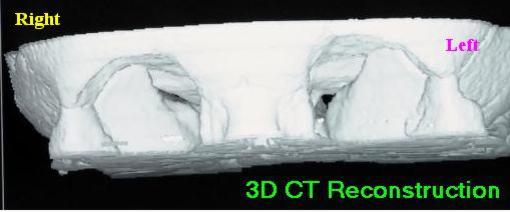
If osteoclasts are not working properly then the canal progressively narrows, putting pressure on the vessels which surround the nerve and carry blood to it. If starved of blood the nerve dies, thins and may then pull clear of the bone. The optic disc at the back of the eye then looks excessively pale when doctors examine the eye using an ophthalmoscope; this is called optic atrophy.
Dense bones in babies with osteopetrosis are clearly visible in the last trimester of life in the womb. It is therefore very likely that damage to the optic nerve probably happens in the womb although it usually will not become apparent until fixing and following should occur 6 or more weeks after birth. As with all nerve damage this process appears to be irreversible, so that once sight is lost it cannot be regained (even after a successful stem marrow transplant). Occasionally it is possible to prevent further visual loss in a child who is just losing vision by a surgical operation called optic nerve decompression but this often appears to be unsuccessful.
Occasional children can develop degeneration of the retina, usually in conjunction with a severe neurological form of the disease called neuronopathic osteopetrosis (see below).
"Large Heads"
The bony thickening is much more severe in the base of the skull than the vault (dome). This causes relative prominence ("bossing") of the forehead, an appearance aggravated by poor growth of the remainder of the body - children with osteopetrosis tend to be relatively short and therefore low on their growth (centile) charts.
Misery / Irritability & Fractures
Like the snuffles described above, misery is not usually listed as a major symptom. However, babies with osteopetrosis are often irritable. This may be due to one of the following:
unrecognised bone fractures
low blood calcium
headache due to increased venous pressure within the skull
central nervous system deterioration in children with neuronopathic osteopetrosis.
Fractures are found in the majority of children if X-rays of all of their bones (a skeletal survey) are carefully examined. Particularly common sites are the long bones of the limbs, the ribs and the acromial processes - small bony projections from the shoulder bone (scapula) (see below).
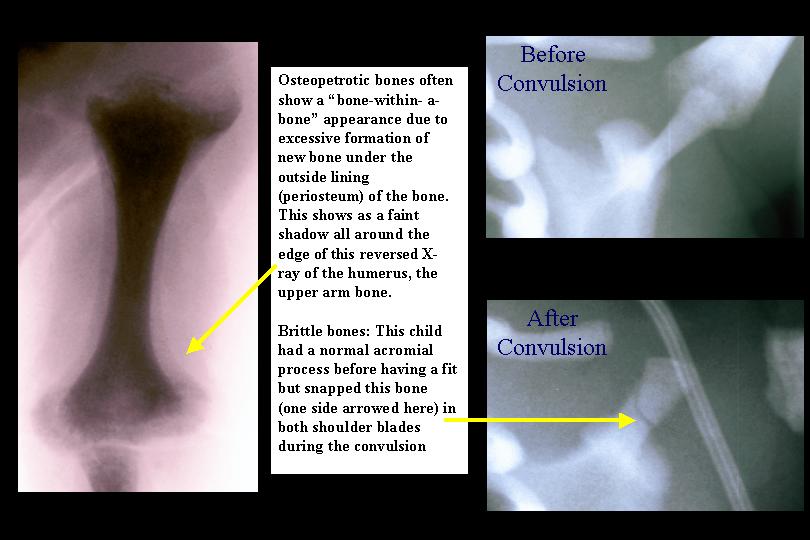
These fractures often occur during passage though the birth canal since osteopetrotic bones are very brittle, resembling solid sticks of chalk.
High venous pressure results from constriction of one of the major veins draining blood from the brain where it passes through a canal called the jugular foramen. Bone compression here increases venous pressure inside the head, causing back pressure which is shown by dilation of veins on the outside of the skull. The typical appearance of a miserable baby with venous dilation is shown below.

High venous blood pressure can affect the absorption of cerebrospinal fluid (CSF) from around the outside of the brain, resulting in a condition called communicating hydrocephalus. Hydrocephalus can cause a bulging fontanelle (soft spot); sometimes this only develops during blood or marrow stem cell transplantation when fluid retention often occurs. This sometimes has to be managed by insertion of a ventriculo-peritoneal shunt which drains CSF from around the brain into the abdominal cavity.
Low Calcium Symptoms (Jitters / Fits)
In the first month of life babies are at a crucial stage with regard to calcium balance, due to rapid bone mineralisation and growth; this can be worsened if food intake is also poor. If calcium levels drop significantly, children become irritable and jittery. At worst they may begin to convulse. It is important to recognise osteopetrosis as a potential cause of neonatal hypocalcaemia and neonatal convulsions occurring in the first month of life. Recognising this association is critical since children diagnosed at this age may reach transplant in time to save their vision.
Blood Problems (Anaemia / Thrombocytopenia)
The blood contains four major subtypes of cell:
* Red blood cells, which carry oxygen around the body; lack of sufficient red blood cells is called anaemia.
* Platelets, small fragments of cells which stick to damaged parts of blood vessels; if their numbers drop we begin to bruise easily or develop minute purple spots (petechiae) in the skin. Lack of platelets is called thrombocytopaenia and can result in severe bleeding.
* Granulocytes or neutrophils, white blood cells whose main purpose is to fight bacteria. A lack of these is termed neutropenia.
* Lymphocytes, another type of white blood cell which help to fight viral infections and to produce antibodies.
In early intrauterine life blood is mostly produced in the liver and spleen ("extramedullary haemopoiesis"). During the second trimester of pregnancy this gradually switches to bone marrow, a process which is usually complete by birth. If this does not occur the liver and spleen stay large (hepatosplenomegaly) and are readily felt in the abdomen.
This different site of blood production causes characteristic appearances when the blood is examined down the microscope (leukoerythroblastic anaemia). The white blood count tends to be high initially but can fall later. Anaemia is common, as is a low platelet count: probably resulting from increased blood destruction by the spleen ("hypersplenism"). This process tends to worsen with age as the spleen increases in size. Eventually regular transfusions of blood and platelets become necessary.
Approximately three quarters of children develop blood problems in the first year of life; a few also develop problems later. Occasional children show normalisation of their liver and spleen size and blood problems, even though their bones remain dense. Those who develop major impairment of their blood count in the first 3 months of life have a poor outlook.
Infection
Low white blood counts due to hypersplenism may predispose to infection. However, there is also some evidence that the bacterial killing mechanism of neutrophils is reduced due to impaired production of superoxides. This is the basis of a treatment based on repeated subcutaneous injections of gamma interferon, which has been reported to reduce infection rates.
Infection is possibly more common in older children and adults, especially osteomyelitis (infection of the bone). Patients with all types of the disease often suffer from delayed eruption of teeth and poor quality teeth which are very prone to decay - this predisposes them to serious osteomyelitis of the jaw.
TREATMENT OF MALIGNANT INFANTILE OSTEOPETROSIS
Prognosis for Untreated Disease
The outlook for children with untreated malignant infantile osteopetrosis is poor as only 30% survive until the age of 6 years and almost all will die without treatment by their mid-teens. Quality of life is often poor as many children are blind, and can later become deaf or develop hydrocephalus. Breathing can be obstructed and some children require insertion of a breathing tube into their trachea called a tracheostomy. Eventually a patient's ability to clear their secretions is reduced and many develop serious chest infection. Progressive enlargement of the spleen causes the blood count to fall which may increase infection risk.
Medical Treatment
There are two main forms of treatment: (a) a variety of medicines and (b) peripheral blood stem cell (PBSC), cord blood or bone marrow transplantation (BMT). The drug treatments are designed to reduce symptoms whereas stem cell transplantation aims to cure the disease; unfortunately this treatment is not thought to be appropriate in adults with autosomal dominant osteopetrosis.
The main drugs which have been used are as follows:
1. Prednisolone
This steroid drug has been used at both high and low doses. The main beneficial effects are on blood consumption by the enlarged spleen so that anaemia and thrombocytopenia may be improved. At high doses reductions in bone density and the size of marrow cavities can occur, probably due to a direct bone thinning effect of steroids. However, these effects are temporary and high dose steroids have many unacceptable side effects. These include increased infection (especially oral thrush), weight gain and swelling of the face, diabetes, high blood pressure and later development of cataracts.
Low dose steroids, however, have far fewer side effects yet may reduce transfusion requirements. They can be considered in a child who is not a candidate for stem cell transplantation.
2. Interferon-(gamma)1b
The use of interferon- comes from the observation that the superoxide system which neutrophils use to kill bacteria is abnormal in many patients with osteopetrosis. This is of interest since osteoclasts and neutrophils are both produced by the same bone marrow precursor (parent) cells. Interferon- is a drug which can stimulate superoxide activity.
Interferon-1b is used as an injection under the skin (subcutaneous) 3 times per week. No serious side effects have been reported. Dr. Lyndon Key and his colleagues described the use of this treatment in 14 children, 11 of whom had received the drug for at least 18 months. They found evidence of bone resorption and increased marrow spaces (reflected by improved red/white cell and platelet counts) in all patients. A substantial reduction in the rate of infections was also reported.
This therapy has been widely trialed, especially in the USA, because it is so much safer than stem cell transplantation. However, many families have eventually opted to stop treatment because of the need for repeated injections and the fact that the disease is only partially alleviated.
3. Calcitriol (1,25 hydroxy vitamin D)
This drug has been given at high dose on the assumption that the osteoclasts in malignant infantile osteopetrosis might be resistant to the usual stimulatory effects of vitamin D. The treatment of two patients has been described: The first showed some responses that suggested that osteoclast activity had increased, but there was only a slight improvement in their clinical problems. The second showed no improvement. This treatment is no longer routinely used.
HAEMOPOIETIC STEM CELL TRANSPLANTATION
Osteoclasts developed from cells which arise from bone marrow stem cells. As a result it is possible to cure the disease by transplantation using blood or marrow stem cells. Originally bone marrow transplants were used but today either bone marrow, cord blood or peripheral blood stem cells are used according to how well matched a donor can be found. The transplant units most experienced in performing these transplants usually try to use either bone marrow from a fully or very closely matched donor or peripheral blood stem cells (PBSC) from a less well matched donor.
Transplants for osteopetrosis are relatively difficult to perform compared to transplants for other non-malignant diseases. The reasons for this are still being explored but these include high rates of a variety of the complications that follows stem cell transplantation. However these problems are being resolved steadily and cure rates are now typically reported to be as high as 80-95%.
It is important to try to identify rapidly the cause of the osteopetrosis since some genetic types of infantile osteopetrosis are not appropriate for transplantation. Transplant is definitely contraindicated in children with OSTM1 neuronopathic and TNFSF11 disease, and careful consideration is required in children with ClCN7 mutations since late neurological disease can occur in this form despite successful transplantation.
Tissue Matching & Donors
The genes which determine a person's tissue type are inherited in two clusters, one inherited from their father and one from their mother. The genes in this unit code for proteins called the human leukocyte antigen (HLA) system, which allow us to recognise our own cells from those of other people and of micro-organisms. Most units currently examine 5 of these, named HLA-A, B-, C-, DR and DQ.
A donor who matches for all 10 of the maternal and paternal proteins is said to be "10 antigen matched" and is often referred to as being "fully matched". It must be stressed that there are other HLA antigens for which matching is not routinely attempted.
By random chance one in four siblings should be a tissue match for a brother or sister requiring a transplant. However the statistical probability is that one of these four children will also have the disease. This reduces the overall chance of any single sibling being a suitable donor (i.e. free of osteopetrosis and tissue matched) to 1 in 5.
A parent's tissue type is usually exactly half-identical (haploidentical) with their child. It is only common to find other relatives who fully or closely match children in families where cousins intermarry, since genes then recirculate within a family.
There are now many registers of volunteer unrelated donors around the world which carry approximate records of the tissue type of the donors. These can be interrogated by computer to find the best potential matches (a "donor search"). After obtaining blood samples from any prospective donors all 10 of the HLA proteins are accurately typed in order to find the best donor.
Venous Access
An indwelling venous catheter with at least two working lumens is required due to the large number of different drugs and blood products administered during transplantation. It should be noted that these catheters, just like any other indwelling artificial device, can become infected and require removal at a later date.
Conditioning Therapy
In order to allow donor stem cells to engraft it is first of all necessary to kill the patient's own bone marrow so as to effectively create a space for the new stem cells. It is also important to suppress the immune system of the patient in order to prevent graft rejection. "Conditioning chemotherapy" is given over approximately one week before the stem cell infusion to achieve both of these aims. The drugs used vary from unit to unit and according to the type of transplant being performed. Commonly these will utilise two or more drugs from this list: busulfan or treosulfan, cyclophosphamide or fludarabine, and thiotepa. Detailed guidelines from the European Blood And Marrow Transplant (EBMT) organisation are given here. These are updated periodically.
There is always a risk that cells in the graft may attack the patient's body, causing a complication called graft versus host disease (GVHD). It is T-lymphocytes (T-cells) in the donor graft which are thought to principally be responsible for causing GVHD. There are many ways of either removing T-cells or of reducing their function and these are very commonly used in transplants for osteopetrosis.
It is possible to give an antibody which kills T-lymphocytes (T-cells) during conditioning therapy. The two major antibodies used are antithymocyte globulin (ATG) or Alemtuzumab. Although these drugs are given before the transplant they persist in the patient's circulation and kill donor T-cells when these are infused into the person's blood. Other immune suppressants are used to prevent GVHD (typically one or more of ciclosporin, steroids, methotrexate, mycophenolate mofetil [MMF], tacrolimus or sirolimus).
Where donors other than sibling donors are being used, the stem cells also may have T-cells removed by physical methods involving microscopically small magnetic beads. These may be used to either prepare relatively pure stem cells (CD34 cell selection) or to try to remove T and B-cells contaminating the graft (TCR alpha beta/CD19 depletion).
A drug called defibrotide may be given to prevent a liver complication called veno-occlusive disease (VOD).
Sometimes it is necessary to give additional stem cell infusions if engraftment is very slow or only partial. These are often termed stem cell boosts. If graft rejection occurs a second transplant may need to be performed after further conditioning chemotherapy.
Stem cell sources
1. Bone Marrow
Bone marrow is collected from the pelvic bones of the donor under general anaesthetic using hollow needles and syringes. The marrow is filtered before being infused into the bloodstream; it is then returned to the patient like a normal blood transfusion.
2. Peripheral Blood Stem Cells
A naturally occurring bone marrow hormone, granulocyte colony stimulating factor (G-CSF), can stimulate bone marrow to release stem cells capable of producing all of the different blood cells (including osteoclasts) into the bloodstream. These are called "peripheral blood stem cells", or PBSC for short. If G-CSF is administered for 4-5 days to a potential donor, PBSCs can then be collected from the blood by running the donor's blood through a machine which collects the stem cells by centrifugation. The remaining blood cells are then returned to the donor. This avoids a general anaesthetic and any discomfort associated with a bone marrow harvest.
A major advantage of PBSCs is that it is possible to collect many more stem cells than are found in a bone marrow collection. Using this much larger dose of cells reduces rejection rates and allows easier administration of stem cell boosts. This technique has been particularly important in development of successful haploidentical transplantation from parents.
3. Cord Blood
Blood drained from the umbilical cord and placenta immediately after the delivery of a baby is very rich in stem cells. Although the actual number of stem cells in cord blood is much lower than from a bone marrow harvest, and many times less than from a PBSC collection, these are often adequate to engraft small children with osteopetrosis.
Cord blood transplantation has several major advantages over BMT or PBSC transplants. Firstly, the cells are available almost immediately, an important consideration in a child whose vision is threatened. Secondly, cord blood donations are less likely to carry the infections which tend to cause serious problems after transplant, e.g cytomegalovirus (CMV), Epstein-Barr virus (EBV: glandular fever virus). Thirdly, they cause relatively less graft versus host disease than equivalently matched bone marrow donations.
At the moment cord blood is used rarely although this is likely to change as the number of available units increases. An EBMT survey conducted of cord blood transplants performed for osteopetrosis up until 2012 showed 54% three year survival for those with the best matched category of cord blood transplants (6/6 matched).
The Effects of Bone Marrow Transplantation on the Disease
Bone clearing after transplantation is demonstrated in these leg X-rays from a child who received an unrelated donor bone marrow transplant.
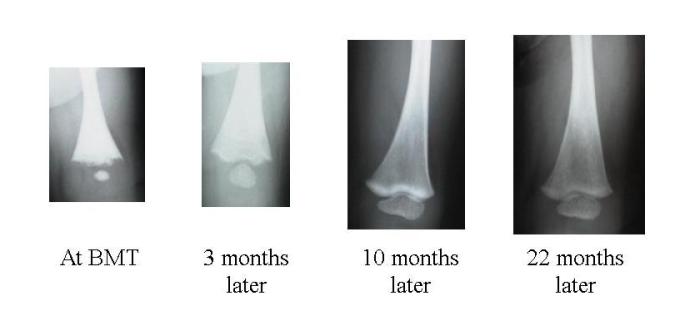
Further overgrowth of the passages in which nerves pass through the skull is prevented by successful transplantation. This usually prevents deafness, hydrocephalus or paralysis of facial muscles. Patients with partial vision at the time of transplant may well retain useful vision or light/dark perception. However, if vision has been completely lost this cannot be regained.
Blood counts normalise after successful transplant although this may take up to one year to be complete.
Liver and spleen enlargement (hepatosplenomegaly) reduces and eventually disappears as bone marrow cavities reform.
Although we would expect catch-up growth once osteoclasts are functioning normally after BMT this does not usually occur. The reason for this is unclear. Most children will therefore grow along a similar growth centile to the one that they were following at the time of transplant.
TRANSPLANT COMPLICATIONS
Although stem cell transplantation is the only curative therapy for malignant infantile osteopetrosis, there are many potential risks. Particular risks are described below:
Convulsions
One of the drugs most commonly used in conditioning therapy before transplantation is called busulfan. A side effect of this drug is fitting (convulsions), and this risk may be aggravated by the use of ciclosporin for GVHD prevention and high blood pressure (which is common during stem cell transplants). Convulsions mostly occur during or soon after busulfan therapy and anti-epileptic drugs such as clonazepam are often used to protect against these.
When fits occur they are usually easy to control and cause no lasting harm. It is rare to see fitting beyond the immediate transplantation period.
Veno-occlusive disease (VOD)
This side effect is also caused by obstruction of the small veins within the liver. It is most common in babies undergoing transplantation and where busulfan is used during conditioning therapy, and is especially common in patients with osteopetrosis. VOD is characterised by enlargement of the liver (hepatomegaly) with pain due to stretching of the capsule around the liver, jaundice (an increased bilirubin level in the blood) and fluid retention with weight gain.
Blood proteins such as albumin leak into the tissues and hold fluid outside the blood system. This fluid can accumulate around the guts (this is called ascites) and lungs (pleural effusions). This side effect develops during the first month after transplant and can vary from being mild to life threatening. Many children will develop mild elevation of their bilirubin level and some weight gain; only a minority will develop severe ascites and pleural effusions.
The drug defibrotide may be used during the early weeks of transplant to try to prevent development VOD, or used for treatment once the complication becomes established. Ursodeoxycholic acid may also be used to increase bile excretion.
Infections
These are one of the most major risks for any patient undergoing BMT. The commonest infections are bacterial infections caused by organisms which usually live harmlessly in the gut or on the skin. These invade once the mouth and gut lining become inflamed due to chemotherapy and the white cell count has fallen very low. They may colonise the plastic of a Hickman catheter and be difficult to eradicate without removing the line. Powerful antibiotic combinations are used at the first sign of fever.
Fungal infections with organisms such as Candida and Aspergillus also pose a threat. Drugs such as fluconazole and itraconazole are used in an effort to prevent infection but other antifungal medication is added if unexplained fevers do not respond to antibiotic therapy.
Other rare organisms which rarely cause infection in health can cause disease after BMT. A good example is Pneumocystis which can cause severe lung infection; this can be effectively prevented by treatment with co-trimoxazole (Septrin).
Viral infections are the most difficult group to combat since few truly effective and non-toxic antiviral drugs exist. The viruses which cause cold sores and chicken pox (Herpes simplex and Varicella zoster respectively) remain resident in our bodies after infection earlier in life and commonly reactivate following stem cell transplantation - producing cold sores/mouth ulcers or shingles. The drug aciclovir is commonly used to prevent this occurring.
Cytomegalovirus (CMV) infection can arise from the body of the patient or the blood cells of their donor and cause serious complications, especially inflammation of the lungs termed pneumonitis. Transplant doctors therefore carefully monitor the CMV status of donors at the time of donor selection and of patients during the early months after transplant. Apart from these resident viruses other common respiratory (e.g. adenovirus, influenza, parainfluenza and RSV) and gut (e.g. rotavirus, Norovirus) viruses can cause also cause infections.
The risk of infection persists long after transplant, especially following unrelated or haploidentical donor transplants. This is aggravated if GVHD develops, necessitating treatment with immune suppressive drugs such as steroids. Spleen function is also reduced by transplantation and many units recommend prolonged or lifelong administration of penicillin to help prevent the infections which the spleen normally protects against.
Graft versus Host Disease (GVHD)
The commonest sign of GVHD is a skin rash which usually develops soon after neutrophil engraftment. GVHD can also affect the liver, causing jaundice and liver dysfunction, and the gut, causing diarrhoea. Usually these symptoms can be controlled using steroid drugs, started at high doses and then wean down to eventual stoppage according to response. If acute GVHD (occurring during the first 3 months after transplant) is not effectively controlled it tends to enter a chronic phase (chronic GVHD) which behaves more like an autoimmune disease. This particularly affects the skin, lungs and immune system.
Hypertension
BMT almost invariably results in fluid retention, especially in the first 1-2 weeks after the transplant. This is aggravated by the use of ciclosporin +/- steroids and other drugs which impair kidney function (e.g. gentamicin, vancomycin, amphotericin) and may result in elevation of blood pressure requiring anti-hypertensive drug therapy. This is usually only a short term problem.
Pulmonary Hypertension
This complication was only first reported in osteopetrosis in 2004 but has come to be seen as an important cause of an excess of lung problems seen in children transplanted for this disease. Previously these had often been ascribed to viral infections or "pneumonitis". The most common presentation is acute breathlessness, reduced blood oxygen levels and brady/tachycardia - usually in the absence of fever, lung crepitations or other evidence of infection. Many patients will respond to treatment with one or more of prostacyclin, sildenafil and defibrotide.
Hypercalcaemia
This occurs in older children after BMT, especially those over the age of 2 years. By this stage the body stores of calcium in a child with osteopetrosis are extremely high. When calcium is mobilised from bone by engrafting donor osteoclasts this can overwhelm the kidney's ability to excrete calcium, causing rapid and dangerous rises in blood calcium. This is most commonly managed with calcium lowering drugs Bisphosphonates, and more recently with Denosumab.
USEFUL REFERENCES
A review of Osteopetrosis published in 2009 can be downloaded from the Orphanet Journal of Rare Diseases
Diagnosis and Management of Osteopetrosis: Consensus Guidelines From the Osteopetrosis Working Group